Final Presentation
Paper Presentation
Rost, Burkard. "Neural networks for protein structure prediction: hype or hit?," 2003. http://cubic.bioc.columbia.edu/papers/2003_rev_nn/paper.pdf
Problem Overview
The protein folding problem - i.e. how to predict a protein's three-dimensional structure from its one-dimensional amino-acid sequence - is often described as the most significant problem remaining in structural molecular biology; to solve the protein folding problem is to break the second half of the genetic code. On a practical level, solving the protein folding problem is the key to rapid progress in the fields of protein engineering and 'rational' drug design. Moreover, as the number of protein sequences is growing much faster than our ability to solve their structures experimentally (e.g. using x-ray crystallography) - creating an ever-widening sequence-structure gap - the pressure to solve the protein folding problem is increasing.
The prediction of a protein's secondary structure - i.e. the formation of regular local structures such as alpha-helices and beta-strands within a single protein sequence - is an essential intermediate step on the way to predicting the full three-dimensional structure of a protein. If the secondary structure of a protein is known, it is possible to derive a comparatively small number of possible tertiary (three-dimensional) structures using knowledge about the ways that secondary structural elements pack.
-- Adrian Shepherd [1]
Introduction
It is apparent that solving the protein folding problem is of paramount concern to molecular biology. Solving this problem would allow for great strides across the board in all of the medical and biological fields of science. Proteins are formed by joining 20 different amino acids into a stretched chain. In water, the chain folds into a unique three-dimensional (3D) structure. In proteins, the sequence of amino acids determines the shape, and the shape determines the function. Thus knowing the structure of a protein is key to understanding and utilizing the protein. The computational effort required to study protein folding is enormous. Using crude workload estimates for a petaflop/second capacity machine leads to an estimate of three years to simulate 100 microseconds of protein folding.
Why Neural Nets?
Because of this, many researchers have been trying to find possible shortcuts into using computational methods to find the structure of proteins. One such option that has shown itself to be viable in the last 20 years has been the neural network. Neural network solutions do not require intensive amounts of computation, and they can be trained to provide extremely accurate (upwards of 70-80% accuracy) results. While the basic methods of using neural networks are all relatively the same, what differs between each method is how biological data can be encoded into the neural network, and how the neural network deals with this biological data.
Neural networks generalise by extracting the underlying physico-chemical principles from the training data. Obviously, this requires a correct training set. Søren Brunak has pioneered the idea to unravel errors in the training set by monitoring samples that could not be learned even when the networks were trained until over-fitting the data. This technique has not only been used successfully to identify errors, and inconsistencies in public data bases, but also to improve the network performance.
Secondary Structures and Neural Nets Examined
So in protein structure prediction with neural networks (or any soft computing technique), there is one common goal: to find a general mapping from the amino acid sequence to the secondary structure. If you don't recall, the secondary structure of a protein consists of the identification of common three-dimensional substructures found within the larger protein structure. These three substructures consists of alpha-helixes (which appear as coils or springs in common 3D representations), beta-sheets (which appear as large flat arrows), and coils (which are simply non-descript threads connecting everything together).
So why not just try and predict the tertiary (or complete 3D structure) instead of trying to predict the secondary structure first? There are a few reasons actually. The first being that the tertiary structure consists of the individual molecules and even atoms of the protein, whereas the secondary structure is merely a rough description of the protein. To predict a 3D structure from the kind of granularity present in tertiary structures represents a significant challenge for neural networks. The atoms would more likely be perceived as noise rather than patterns. Secondary structures avoid this because a secondary structure is only concerned with general descriptions of a protein's substructures. It is known that these substructures often occur in patterns and thus a neural network solution should be viable due to it's inherit pattern matching and recognition abilities. Another added complexity to predicting tertiary structures is that the three-dimensional structure of a protein is position and orientation independent. So training a neural network to recognize this independence would add another complex element to training. Secondary structures do not suffer from this complexity because a secondary structure is merely a chain of two-dimensional descriptors.
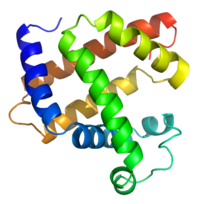
We're after the tertiary structure in the long run, so how does finding the secondary structure actually help? As it turns out, once a secondary structure is known, it is rather easy to get a good estimate of a low resolution tertiary structure. By low resolution I am refering to the granularity of the molecules and atoms. At the lowest resolution you have just the three dimensional shape of the secondary structure (fig. 1) and at the highest resolution you have the individual atoms and molecules (fig. 2). From the figures it's easy to see how pattern recognition with a secondary structure would be much much easier than with the tertiary structure.
 |
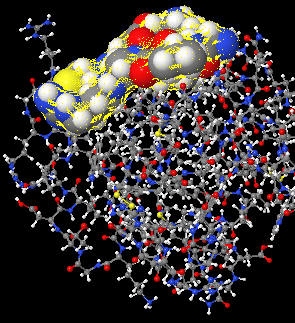 |
| Figure 1: Secondary structure in 3D. | Figure 2: High resolution tertiary structure. |
Methods
There have been a number of different methods developed over the 30 or so years neural nets have been employed to solve the protein folding problem. However, all of the solutions are fundamentally the same and even the current generation of applications show a remarkable similarity to the applications of the past. All solutions are essentially neural networks (almost exclusively your normal back-prop neural net), examining a window of the amino acid chain. The output of the network is one of three outputs corresponding to the three different components of secondary structures (alpha-helix, beta-sheets, and coils).
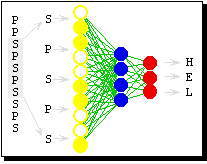 |
Figure 3: Simple neural network for secondary structure prediction. For simplification the protein sequence given consists of two amino acid types (S and P). The protein sequence is translated into patterns by shifting a window of w adjacent residues (shown w = 5; typical values in practice are w = 13-21) through the protein. The output of the network is uniquely determined. Suppose the output would be: 0.2, 0.4, 0.5 for the three output states (H, E, L). For known examples the desired output is also known (1, 0, 0 if the central residue is in a helix). Consequently, the network error is given by the difference between actual network output and desired output. The only free variables are the connections. Training or learning means changing the connections such that the error decreases for the given examples. A training set typically comprises some 30,000 examples. If training is successful, the patterns are correctly classified. But how can new patterns be classified correctly? The hope is that the network succeeds in extracting general rules by the classification of the training patterns. The generalisation ability is checked by another set of test samples for which the mapping of sequence window to secondary structure is known as well. Sufficient testing is crucial and has to meet two requirements. First, any significant sequence similarity between test and training set has to be removed. Second, evaluations of expected prediction accuracy have to be based on a sufficient number of test proteins (> 100). --Burkhard Rost [2] |
In the beginning all neural network solutions were essentially black boxes. And with no biological knowledge employed at all, such black boxes could attain a prediction accuracy of around 60%. At the time (late 80s and early 90s), This was similar to the best methods 30 years of research had resulted in by the end of the 80's. So it's apparent that the pattern recognition abilities could be a viable option in solving this problem. As the years went on such neural network applications went on to achieve prediction accuracies >80%. These improvements in accuracy are almost solely the product of the encoding biological data into the neural network and training sets. The papers surveyed represent all of the different facits of biological information that can be used to increase the neural network's ability to see and match patterns in the amino acid sequence.
Starting with the first papers that was published in 1993, "Peptides Secondary Structure Prediction with Neural Networks: A Criterion for Building Appropriate Learning Sets" and "Protein Folding Analysis by an Artifical Neural Network Approach," a typical black box method was used. What was different was the method in which they chose their training sets. In the case of neural networks, they tend to recognize the alpha-helix pattern to a higher degree of accuracy than any other secondary substructure. They believe this is caused by the homologies of the protein. So to counteract the type of overtraining on alpha-helixes that can occur, they opt to employ a "weak homologies" method. That is, they will build up their training sets with increasing percentages of alpha-helixes. In their study they used a total of seven different training sets. The first set had <10% alpha helixes, and each subsequent training set increased in the percentage of alpha helixes with the final training at >56% alpha-helixes. They were hoping to build up a better, more generalized bond between the secondary structural patterns and the protein's amino acid chain.
Their results showed much promise. Their algorithm performed much better than most of the competition in protein categories that did not include high percentages of alpha-helixes. However, one algorithm performed consistently better on protein sets with high alpha-helix percentages, the Kneller method. The Kneller method also includes tertiary structural class information along with the amino acid chains. The paper supposes that the this method will perform much better on only certain types of proteins (either mostly all alpha-helixes or all beta-sheets). The tertiary class information does not carry any information related to the mixture and interaction of the different secondary structures. However, their method is not so specialized and they claim it has no such restrictions and that it carries information on all three secondary structure types. Also, their training sets were only limited to about 1300 amino acids, which is only about 10-15 proteins each. If their training sets had been larger, >100 proteins each, then they believe they would have gotten results around the 70% accuracy range.
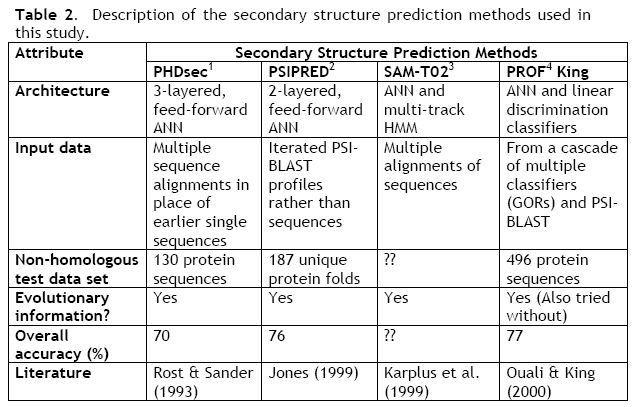
The next paper, "Artificial Neural Networks and Hidden Markov Models for Predicting the Protein Structures: The Secondary Structure Prediction in Caspases," attempts to test and compare the current state of the art on protein prediction. Right now the current leading applications employ such different methods that they are at least slightly specialized towards certain types of proteins. So the goal of the paper was to find a good method for comparing the different applications; the workings of each application are discussed and tested. Note: At the core of all of the current methods is a back propogating neural network. To test the different methods a number of proteins were used from the Caspases family of proteins. I believe they were chosen because the Caspases is a rather large family, so you can have proteins from the same family that are very similar, and proteins that are very different (but since they're in the same family there will be some kind of relationship between them all).
The first method tested was PHDsec. PHDsec is famous for being the firsth method to systematically use multiple sequence alignments for training the neural network. What this means is that the neural networks would take advantage of biological evolutionary data. And using this data it was able to achieve up to 10% gains of accuracy compared to other methods being used at the time. Some residues can be replaced by others without changing structure. However, not every amino acid can be replaced by any other. In just one evolutionary step (the replacement of an amino acid), the protein can become unstable and collapse. So what multiple sequence alignments mean is that the program, in this case PHDsec, will do a look up on different proteins that are evolutionary similar (i.e. they have similar amino acid sequences with few replacements), and will then compile the results and feed it into the neural network. Figure 4 shows this in more detail.
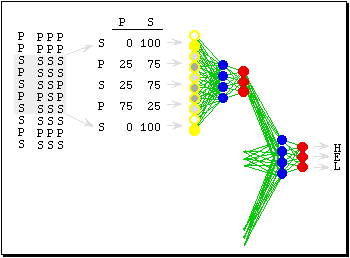 |
Figure 4:Fig. 5. Feeding evolutionary information into a neural network system. (1) A sequence family is aligned (shown are the sequence of unknown structure and three aligned relatives). (2) For each sequence position of profile is compiled that gives the percentage of S, or P in the alignment (shown in centre for window of five adjacent residues). (3) Instead of using binary input units (0 or 1), now the profile is fed into the first neural network. (4) Finally, the output is again fed into a second level network. --Burkhard Rost [2] |
The addition of evolutionary information was a scientific breakthrough, and this method is still one of, if not the most, important step in getting past the 70% accuracy barrier. "The major limitation of PHDsec is that multiple sequence alignment is time consuming and it is difficult to move the PHD server to another site. The alignments usually correlate to sequence diversity. The diversity of the training set influences the prediction accuracy. This method uses only one line of evidence from the alignments to train the networks" --Anekonda [5].
The second method used was PSIPRED. "This was the first method to take advantage of position-specific scoring matrices generated by the powerful PSI-BLAST search algorithm for predicitng protein secondary structures" --Anekonda [5]. This algorithm is the key to PSIPRED's accuracy. Using the PSI-BLAST search the accuracy rose from 70 to above 76%. The PSI-BLAST search is a very good database searching algorithm that is able to find proteins with greater local alignment reliability than traditional searches. Like PHDsec, this method can also handle long distance interactoins. The paper attributes this method's greater accuracy to homologous tertiary structures rather than actually having better used all of its available data.
The next method examined was the SAM-T02 method. SAM-T02 makes use of both neural networks and multi-track hidden Markov models (HMM). While the HMM can't handle finding long distance correlations, the neural network can, and so SAM-T02 makes use of this hybrid architecture. When disadvantage is that the architecture is so complex that meaningful biological rules cannot be extracted from the network.
The last method used is PROF King. PROF King uses many different theories and algorithms including the 7 GOR methods and the neural networks with multiple sequence alignments. PROF King implements a huge ruleset, with it's rules being derived from the many many theories and algorithms that are out there. Combining this many different architectures makes the calculations slow and convoluted, and it also makes any kind of biological data that could be gleaned from studying the network. Table 2 shows the descriptions and the overall results of the tests.
Remarks
Right now the state of the art of secondary structure predictors for proteins is at about 70-80% accuracy depending on the training sets and the family of proteins that get fed into the neural network. This is definately an amazing feat considering how long it would take the world's fastest super computers to come up with a prediction. However, 80% is still not 90%, and to make an even bigger impact on the scientific community neural network solutions need to pass the 90% barrier. I agree with the authors of the papers below in that the next advancements in neural networks as secondary structure predictors will be in encoding even more biological information into the neural networks. I did not see any papers that have done this yet, but I think it may be interested to include partially folded proteins, and maybe train the network to go through the whole folding process. Or possibly build up the training data from partially folded proteins (like in the first paper how they built up from the percentages of alpha-helixes). From these partially folded proteins you may be able to gleem some light on the patterns and rulesets that govern proteins without the added complexity of worrying about 100 other amino acids. And giving the neural net time to learn from these basic sets of proteins that haven't finished learning it may build a better intuition on the patterns of the more complex, completely folded protein.
I'd also like to see if more experimentation on the neural net side would yield better or worse results. Every single method I've looked at uses a back-prop neural net with basic sigmoid functions at its core. It may be interesting to see how radial basis functions may work, or clustering, or ART, etc.
References
- Shepherd, Adrian. "Protein Secondary Structure Prediction with Neural Networks:
A Tutorial," 1999. http://www.biochem.ucl.ac.uk/~shepherd/sspred_tutorial/ss-index.html - Rost, Burkard. "Neural networks for protein structure prediction: hype or hit?," 2003. http://cubic.bioc.columbia.edu/papers/2003_rev_nn/paper.pdf
- R. Sacile and C. Ruggiero. "Protein Folding Analysis by an Artifical Neural Network Approach," 1993. http://ieeexplore.ieee.org/iel5/7705/21095/00979269.pdf?arnumber=979269
- R. Sacile and C. Ruggiero, G. Rauch. "Peptides Secondary Structure Prediction with Neural Networks: A Criterion for Building Appropriate Learning Sets," 1993. http://ieeexplore.ieee.org/iel5/10/6298/00245628.pdf
- Anekonda, Thimmappa. "Artificial Neural Networks and Hidden Markov Models for
Predicting the Protein Structures: The Secondary Structure
Prediction in Caspases," 2002. http://cmgm.stanford.edu/biochem218/Projects%202002/Anekonda.pdf - J. Meiler, D. Baker. "Coupled prediction of protein secondary and tertiary structure," 2003. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pubmed&pubmedid=14528006